Direct prediction of residual dipolar couplings of small molecules in a stretched gel by stochastic molecular dynamics simulations
22-Jan-2015
Magnetic Resonance in Chemistry, 2015, DOI: 10.1002/mrc.4181, Volume 53, Issue 3, pages 213–217 published on 22.01.2015
Magnetic Resonance in Chemistry, online article
Magnetic Resonance in Chemistry, online article
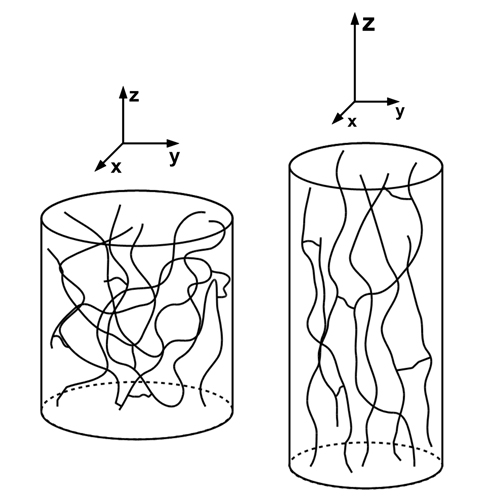
Residual dipolar couplings are highly useful NMR parameters for calculating and refining molecular structures, dynamics, and interactions. For some applications, however, it is inevitable that the preferred orientation of a molecule in an alignment medium is calculated a priori. Several methods have been developed to predict molecular orientations and residual dipolar couplings. Being beneficial for macromolecules and selected small-molecule applications, such approaches lack sufficient accuracy for a large number of organic compounds for which the fine structure and eventually the flexibility of all involved molecules have to be considered or are limited to specific, well-studied liquid crystals. We introduce a simplified model for detailed all-atom molecular dynamics calculations with a polymer strand lined up along the principal axis as a new approach to simulate the preferred orientation of small to medium-sized solutes in polymer-based, gel-type alignment media. As is shown by a first example of strychnine in a polystyrene/CDCl3 gel, the simulations potentially enable the accurate prediction of residual dipolar couplings taking into account structural details and dynamic averaging effects of both the polymer and the solute.