Common variants in the HLA-DQ region confer susceptibility to idiopathic achalasia
06-Jul-2014
Nature Genetics, 2014, doi:10.1038/ng.3029, 46, 901–904 published on 06.07.2014
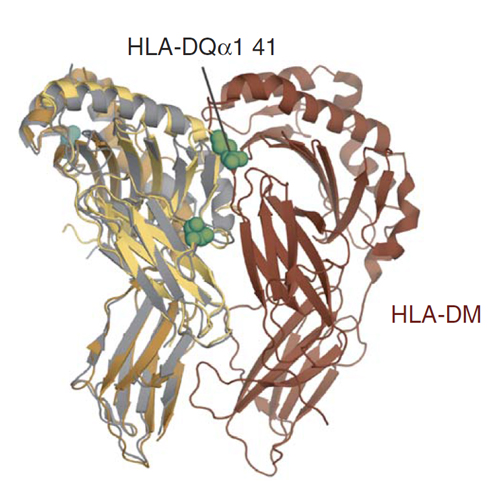
Idiopathic achalasia is characterized by a failure of the lower esophageal sphincter to relax due to a loss of neurons in the myenteric plexu. This ultimately leads to massive dilatation and an irreversibly impaired megaesophagus. We performed a genetic association study in 1,068 achalasia cases and 4,242 controls and fine-mapped a strong MHC association signal by imputing classical HLA haplotypes and amino acid polymorphisms. An eight-residue insertion at position 227–234 in the cytoplasmic tail of HLA-DQβ1 (encoded by HLA-DQB1*05:03 and HLA-DQB1*06:01) confers the strongest risk for achalasia (P = 1.73 × 10−19). In addition, two amino acid substitutions in the extracellular domain of HLA-DQα1 at position 41 (lysine encoded by HLA-DQA1*01:03; P = 5.60 × 10−10) and of HLA-DQβ1 at position 45 (glutamic acid encoded by HLA-DQB1*03:01 and HLA-DQB1*03:04; P = 1.20 × 10−9) independently confer achalasia risk. Our study implies that immune-mediated processes are involved in the pathophysiology of achalasia.