A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease
09-Feb-2015
Nature Medicine, 2015, doi:10.1038/nm.3776, 21, 276–280 published on 09.02.2015
Nature Medicine, online article
Nature Medicine, online article
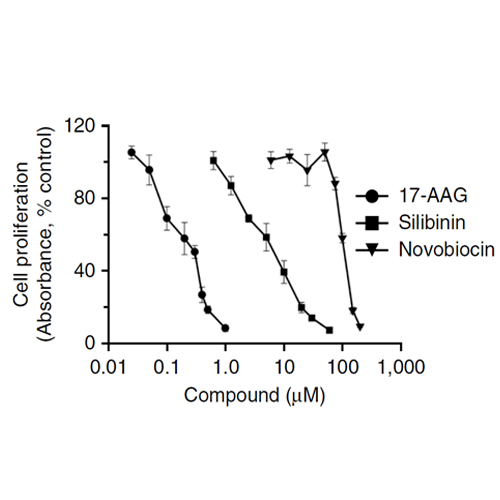
One function of the glucocorticoid receptor (GR) in corticotroph cells is to suppress the transcription of the gene encoding proopiomelanocortin (POMC), the precursor of the stress hormone adrenocorticotropin (ACTH). Cushing disease is a neuroendocrine condition caused by partially glucocorticoid-resistant corticotroph adenomas that excessively secrete ACTH, which leads to hypercortisolism. Mutations that impair GR function explain glucocorticoid resistance only in sporadic cases. However, the proper folding of GR depends on direct interactions with the chaperone heat shock protein 90 (HSP90). We show here that corticotroph adenomas overexpress HSP90 compared to the normal pituitary. N- and C-terminal HSP90 inhibitors act at different steps of the HSP90 catalytic cycle to regulate corticotroph cell proliferation and GR transcriptional activity. C-terminal inhibitors cause the release of mature GR from HSP90, which promotes its exit from the chaperone cycle and potentiates its transcriptional activity in a corticotroph cell line and in primary cultures of human corticotroph adenomas. In an allograft mouse model, the C-terminal HSP90 inhibitor silibinin showed anti-tumorigenic effects, partially reverted hormonal alterations, and alleviated symptoms of Cushing disease. These results suggest that the pathogenesis of Cushing disease caused by overexpression of heat shock proteins and consequently misregulated GR sensitivity may be overcome pharmacologically with an appropriate HSP90 inhibitor.