
Potent Proteasome Inhibitors Derived from the Unnatural cis-Cyclopropane Isomer of Belactosin A: Synthesis, Biological Activity, and Mode of Action
29-Mar-2013
J. Med. Chem., 2013, DOI: 10.1021/jm4002296, 56 (9), pp 3689–3700 published on 29.03.2013
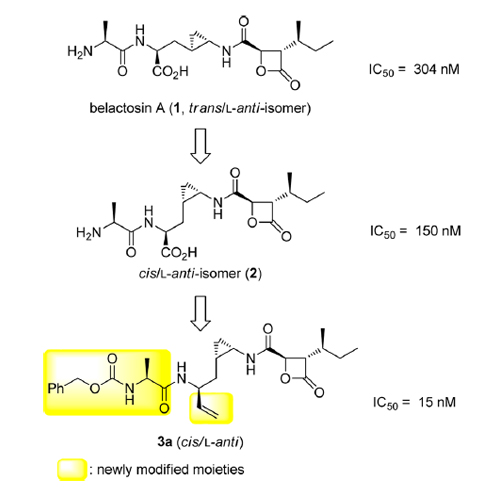
The natural product belactosin A (1) with a trans-cyclopropane structure is a useful prototype compound for developing potent proteasome (core particle, CP) inhibitors. To date, 1 and its analogues are the only CP ligands that bind to both the nonprimed S1 pocket as well as the primed substrate binding channel; however, these molecules harbor a high IC50 value of more than 1 μM. We have performed structure–activity relationship studies, thereby elucidating unnatural cis-cyclopropane derivatives of 1 that exhibit high potency to primarily block the chymotrypsin-like active site of the human constitutive (cCP) and immunoproteasome (iCP). The most active compound 3e reversibly inhibits cCP and iCP similarly with an IC50 of 5.7 nM. X-ray crystallographic analysis of the yeast proteasome in complex with 3e revealed that the ligand is accommodated predominantly into the primed substrate binding channel and covalently binds to the active site threonine residue via its β-lactone ring-opening.