Structural, Biochemical, and Computational Studies Reveal the Mechanism of Selective Aldehyde Dehydrogenase 1A1 Inhibition by Cytotoxic Duocarmycin Analogues
16-Sep-2015
Angew. Chem. Int. Ed., Volume 127, Issue 46, Pages 13754–13758, DOI: 10.1002/anie.201505749
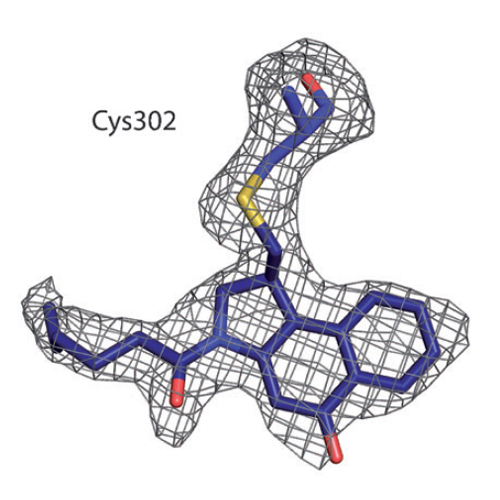
Analogues of the natural product duocarmycin bearing an indole moiety were shown to bind aldehyde dehydrogenase 1A1 (ALDH1A1) in addition to DNA, while derivatives without the indole solely addressed the ALDH1A1 protein. The molecular mechanism of selective ALDH1A1 inhibition by duocarmycin analogues was unraveled through cocrystallization, mutational studies, and molecular dynamics simulations. The structure of the complex shows the compound embedded in a hydrophobic pocket, where it is stabilized by several crucial π-stacking and van der Waals interactions. This binding mode positions the cyclopropyl electrophile for nucleophilic attack by the noncatalytic residue Cys302, thereby resulting in covalent attachment, steric occlusion of the active site, and inhibition of catalysis. The selectivity of duocarmycin analogues for ALDH1A1 is unique, since only minor alterations in the sequence of closely related protein isoforms restrict compound accessibility.